Dr. Lin Recent Publications
- Connected Chromatin Amplifies Acetylation-modulated Nucleosome Interactions. R Li, X Lin. Biochemistry, 2025. DOI: 10.1021/acs.biochem.4c00647.

Abstract: Histone acetylation is a key regulatory post-translational modification closely associated with gene transcription. In particular, H4K16 acetylation (H4K16ac) is a crucial gene activation marker that induces an open chromatin configuration. While previous studies have explored the effects of H4K16ac on nucleosome interactions, how this local modification affects higher-order chromatin organization remains unclear. To bridge the chemical modifications of these histone tail lysine residues to global chromatin structure, we utilized a residue-resolution coarse-grained chromatin model and enhanced sampling techniques to simulate charge-neutralization effects of histone acetylation on nucleosome stability, internucleosome interactions, and higher-order chromatin structure. Our simulations reveal that H4K16ac stabilizes a single nucleosome due to the reduced entropic contribution of histone tails during DNA unwrapping. In addition, acetylation modestly weakens internucleosome interactions by diminishing contacts between histone tails, DNA, and nucleosome acidic patches. These weakened interactions are amplified when nucleosomes are connected by linker DNA, where increases in linker DNA entry-exit angles lead to significant chromatin destacking and decompaction, exposing nucleosomes to transcriptional activity. Our findings suggest that the geometric constraint imposed by chromatin DNA plays a critical role in driving chromatin structural reorganization upon post-translational modifications.
- Interpretable Protein-DNA Interactions Captured by Structure-based Optimization. Y Zhang, I Silvernail, Z Lin, X Lin. bioRxiv, 2024.05. 26.595895.
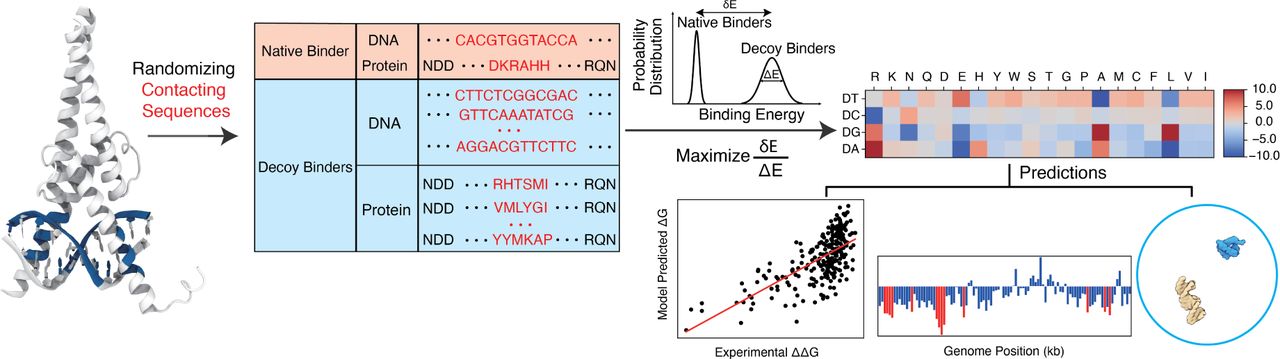
Abstract: Sequence-specific DNA recognition underlies essential processes in gene regulation, yet predictive methods for simultaneous prediction of genome-wide DNA recognition sites and their binding affinity remain lacking. Here, we present IDEA, an interpretable residue-level biophysical model capable of predicting binding sites and strengths of DNA-binding proteins across the genome. By leveraging the sequence-structure relationship from known protein-DNA complexes, IDEA learns an energy model enabling direct interpretation of physicochemical interactions among individual amino acids and nucleotides. Using transcription factors as examples, we demonstrate that this energy model accurately predicts genomic DNA recognition sites and their binding strengths. Additionally, the IDEA model is integrated into a coarse-grained simulation framework that accurately captures the absolute protein-DNA binding free energies. Overall, IDEA provides an integrated computational platform alleviating experimental costs and biases in assessing DNA recognition and can be utilized for mechanistic studies of various DNA-recognition processes.
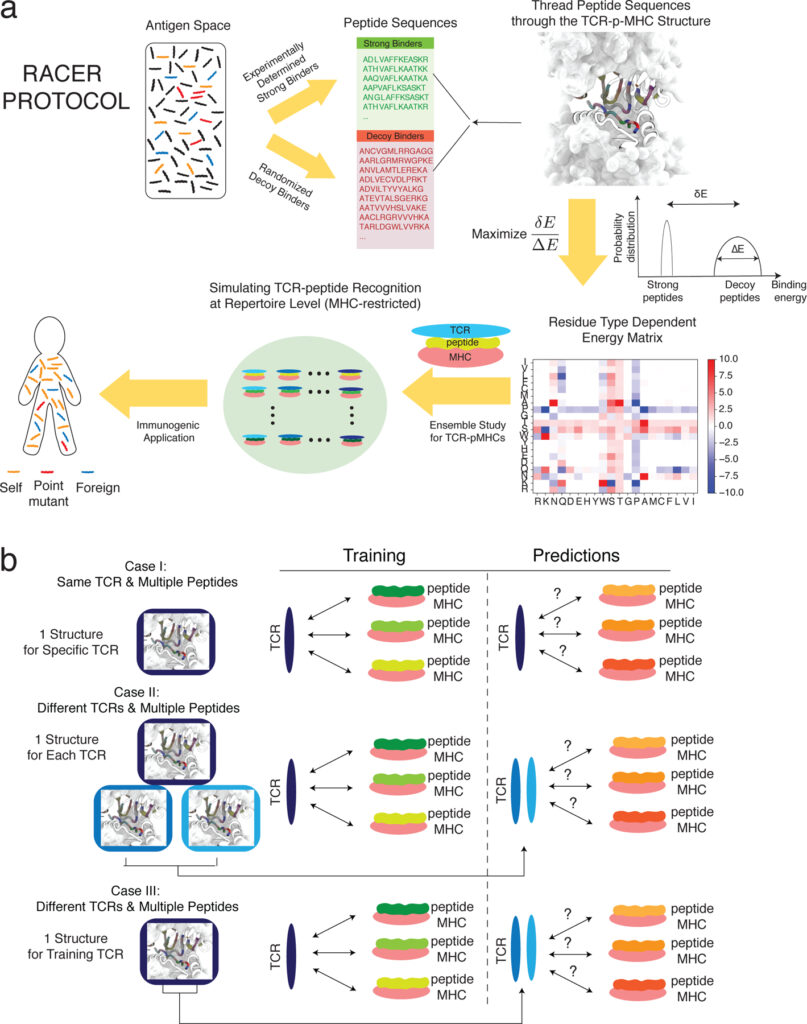
- RACER-m leverages structural features for sparse T Cell specificity prediction. A Wang, X Lin, KN Chau, JN Onuchic, H Levine, JT George. Science Advances 10 (20), eadl0161, 2024.
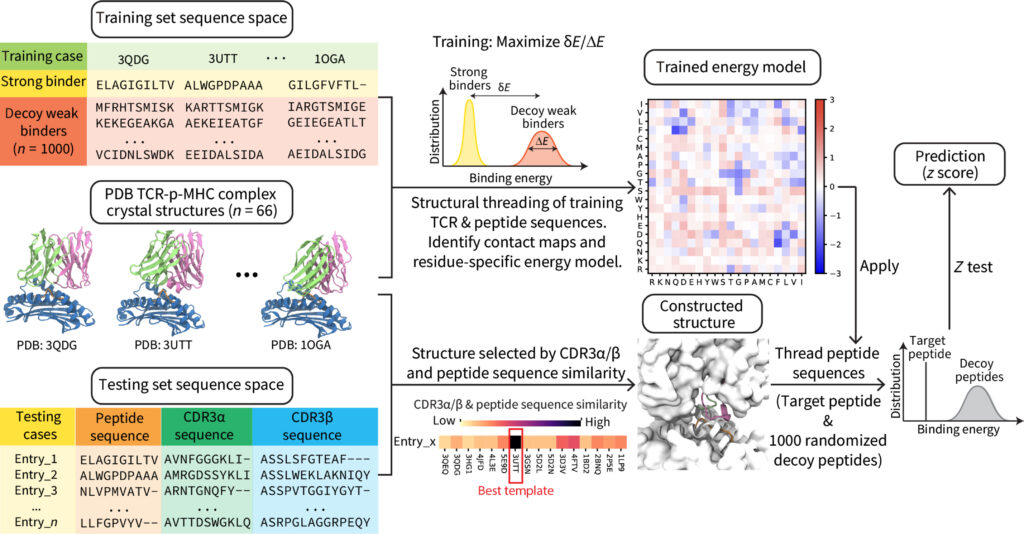
Abstract: Reliable prediction of T cell specificity against antigenic signatures is a formidable task, complicated by the immense diversity of T cell receptor and antigen sequence space and the resulting limited availability of training sets for inferential models. Recent modeling efforts have demonstrated the advantage of incorporating structural information to overcome the need for extensive training sequence data, yet disentangling the heterogeneous TCR-antigen interface to accurately predict MHC-allele-restricted TCR-peptide interactions has remained challenging. Here, we present RACER-m, a coarse-grained structural model leveraging key biophysical information from the diversity of publicly available TCR-antigen crystal structures. Explicit inclusion of structural content substantially reduces the required number of training examples and maintains reliable predictions of TCR-recognition specificity and sensitivity across diverse biological contexts. Our model capably identifies biophysically meaningful point-mutant peptides that affect binding affinity, distinguishing its ability in predicting TCR specificity of point-mutants from alternative sequence-based methods. Its application is broadly applicable to studies involving both closely related and structurally diverse TCR-peptide pairs.
- Explicit ion modeling predicts physicochemical interactions for chromatin organization. X Lin, B Zhang. eLife 12, RP90073, 2024.
Abstract:  Molecular mechanisms that dictate chromatin organization in vivo are under active investigation, and the extent to which intrinsic interactions contribute to this process remains debatable. A central quantity for evaluating their contribution is the strength of nucleosome-nucleosome binding, which previous experiments have estimated to range from 2 to 14 kBT. We introduce an explicit ion model to dramatically enhance the accuracy of residue-level coarse-grained modeling approaches across a wide range of ionic concentrations. This model allows for de novo predictions of chromatin organization and remains computationally efficient, enabling large-scale conformational sampling for free energy calculations. It reproduces the energetics of protein-DNA binding and unwinding of single nucleosomal DNA, and resolves the differential impact of mono- and divalent ions on chromatin conformations. Moreover, we showed that the model can reconcile various experiments on quantifying nucleosomal interactions, providing an explanation for the large discrepancy between existing estimations. We predict the interaction strength at physiological conditions to be 9 kBT, a value that is nonetheless sensitive to DNA linker length and the presence of linker histones. Our study strongly supports the contribution of physicochemical interactions to the phase behavior of chromatin aggregates and chromatin organization inside the nucleus.
Molecular mechanisms that dictate chromatin organization in vivo are under active investigation, and the extent to which intrinsic interactions contribute to this process remains debatable. A central quantity for evaluating their contribution is the strength of nucleosome-nucleosome binding, which previous experiments have estimated to range from 2 to 14 kBT. We introduce an explicit ion model to dramatically enhance the accuracy of residue-level coarse-grained modeling approaches across a wide range of ionic concentrations. This model allows for de novo predictions of chromatin organization and remains computationally efficient, enabling large-scale conformational sampling for free energy calculations. It reproduces the energetics of protein-DNA binding and unwinding of single nucleosomal DNA, and resolves the differential impact of mono- and divalent ions on chromatin conformations. Moreover, we showed that the model can reconcile various experiments on quantifying nucleosomal interactions, providing an explanation for the large discrepancy between existing estimations. We predict the interaction strength at physiological conditions to be 9 kBT, a value that is nonetheless sensitive to DNA linker length and the presence of linker histones. Our study strongly supports the contribution of physicochemical interactions to the phase behavior of chromatin aggregates and chromatin organization inside the nucleus.
- Chromatin fiber breaks into clutches under tension and crowding. S Liu, X Lin, B Zhang. Nucleic Acids Research 50 (17), 9738-9747, 2022.
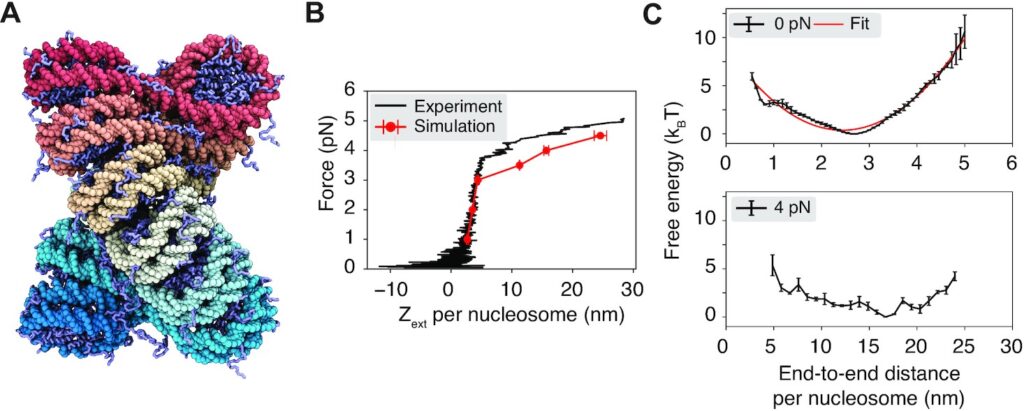
Abstract: The arrangement of nucleosomes inside chromatin is of extensive interest. While in vitro experiments have revealed the formation of 30 nm fibers, most in vivo studies have failed to confirm their presence in cell nuclei. To reconcile the diverging experimental findings, we characterized chromatin organization using a residue-level coarse-grained model. The computed force–extension curve matches well with measurements from single-molecule experiments. Notably, we found that a dodeca-nucleosome in the two-helix zigzag conformation breaks into structures with nucleosome clutches and a mix of trimers and tetramers under tension. Such unfolded configurations can also be stabilized through trans interactions with other chromatin chains. Our study suggests that unfolding from chromatin fibers could contribute to the irregularity of in vivo chromatin configurations. We further revealed that chromatin segments with fibril or clutch structures engaged in distinct binding modes and discussed the implications of these inter-chain interactions for a potential sol–gel phase transition.
- Rapid assessment of T-cell receptor specificity of the immune repertoire. Xingcheng Lin, Jason T George, Nicholas P Schafer, Kevin Ng Chau, Michael E Birnbaum, Cecilia Clementi, José N Onuchic, Herbert Levine. Nature Computational Science 1 (5), 362-373, 2021.
Abstract: Accurate assessment of T-cell-receptor (TCR)–antigen specificity across the whole immune repertoire lies at the heart of improved cancer immunotherapy, but predictive models capable of high-throughput assessment of TCR–peptide pairs are lacking. Recent advances in deep sequencing and crystallography have enriched the data available for studying TCR–peptide systems. Here, we introduce RACER, a pairwise energy model capable of rapid assessment of TCR–peptide affinity for entire immune repertoires. RACER applies supervised machine learning to efficiently and accurately resolve strong TCR–peptide binding pairs from weak ones. The trained parameters further enable a physical interpretation of interacting patterns encoded in each TCR–peptide system. When applied to simulate thymic selection of a major-histocompatibility-complex (MHC)-restricted T-cell repertoire, RACER accurately estimates recognition rates for tumor-associated neoantigens and foreign peptides, thus demonstrating its utility in helping address the computational challenge of reliably identifying properties of tumor antigen-specific T-cells at the level of an individual patient’s immune repertoire.